







Proteomes 2024, 12(2), 15; https://doi.org/10.3390/proteomes12020015 - 10 May 2024
Abstract
One of the human proteome puzzles is an imbalance between the theoretically calculated and experimentally measured amounts of proteoforms. Considering the possibility of combinations of different post-translational modifications (PTMs), the quantity of possible proteoforms is huge. An estimation gives more than a million
[...] Read more.
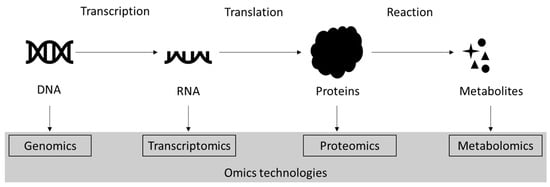
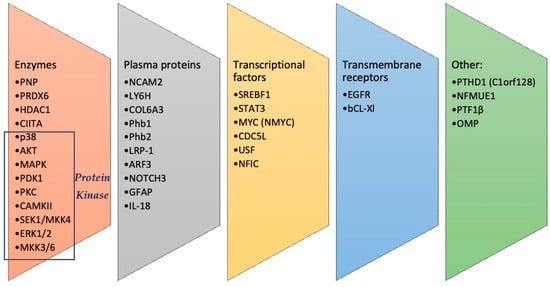
One of the human proteome puzzles is an imbalance between the theoretically calculated and experimentally measured amounts of proteoforms. Considering the possibility of combinations of different post-translational modifications (PTMs), the quantity of possible proteoforms is huge. An estimation gives more than a million different proteoforms in each cell type. But, it seems that there is strict control over the production and maintenance of PTMs. Although the potential complexity of proteoforms due to PTMs is tremendous, available information indicates that only a small part of it is being implemented. As a result, a protein could have many proteoforms according to the number of modification sites, but because of different systems of personal regulation, the profile of PTMs for a given protein in each organism is slightly different.
Full article
(This article belongs to the Special Issue 10th Anniversary of Proteomes—Reviewing the Progress and Prospects of Proteomics)
►
Show Figures

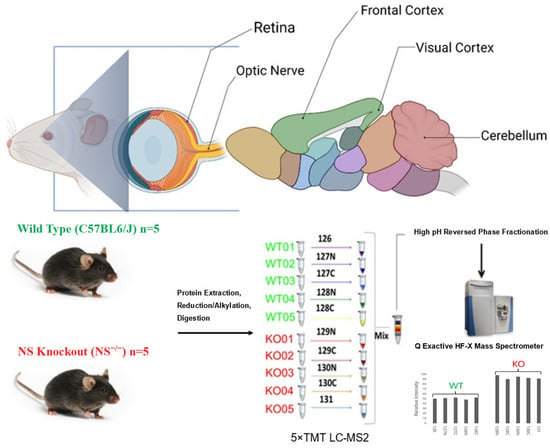
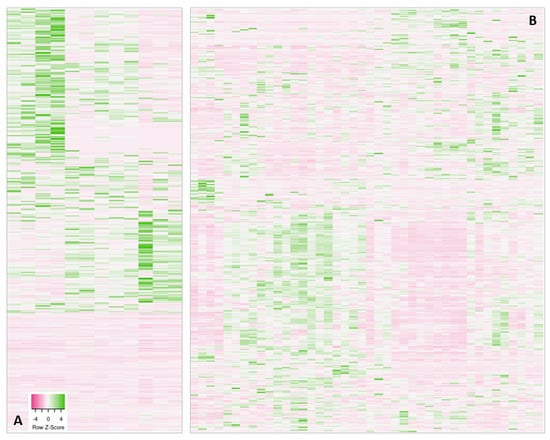
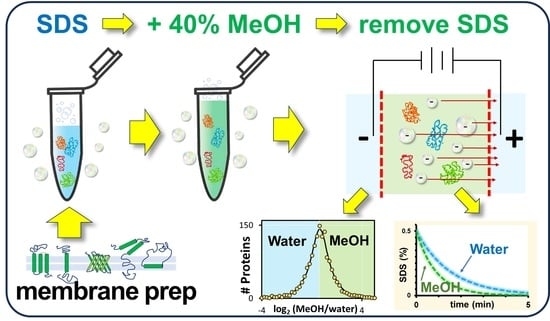
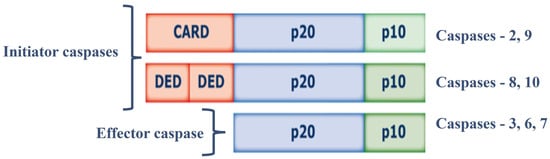
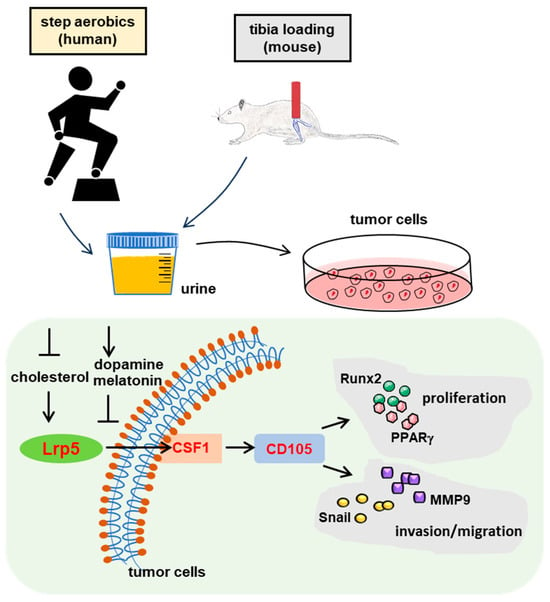
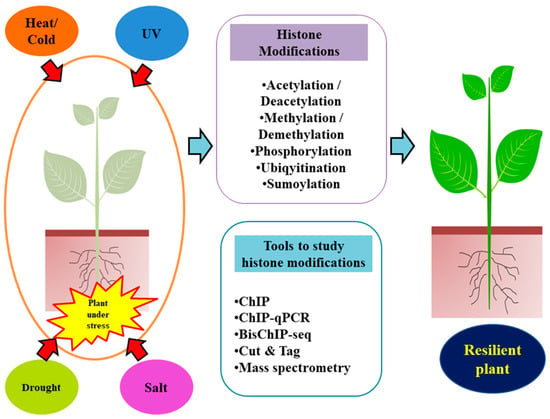
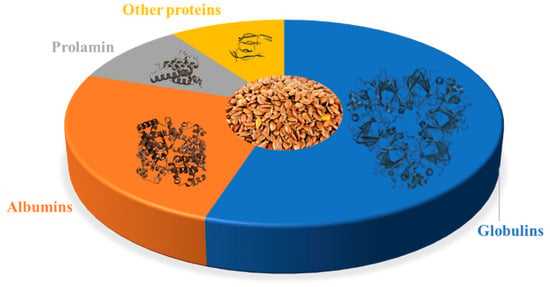
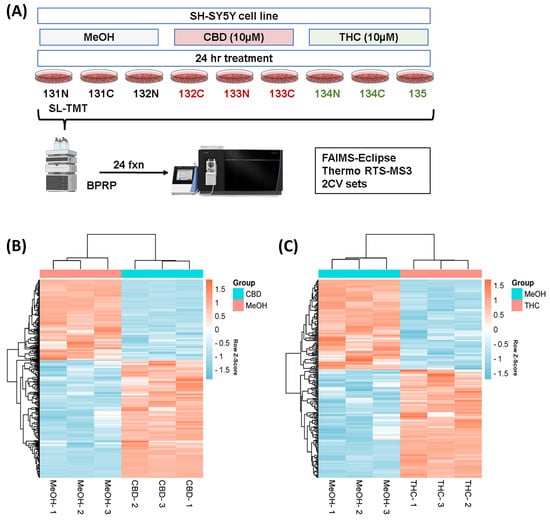
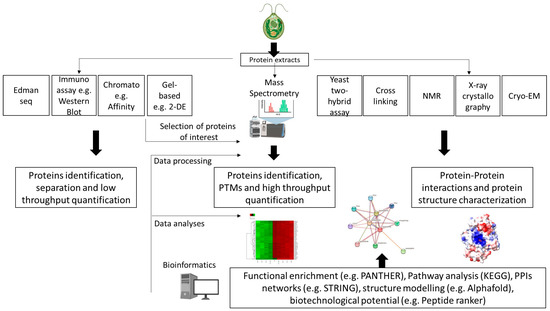
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
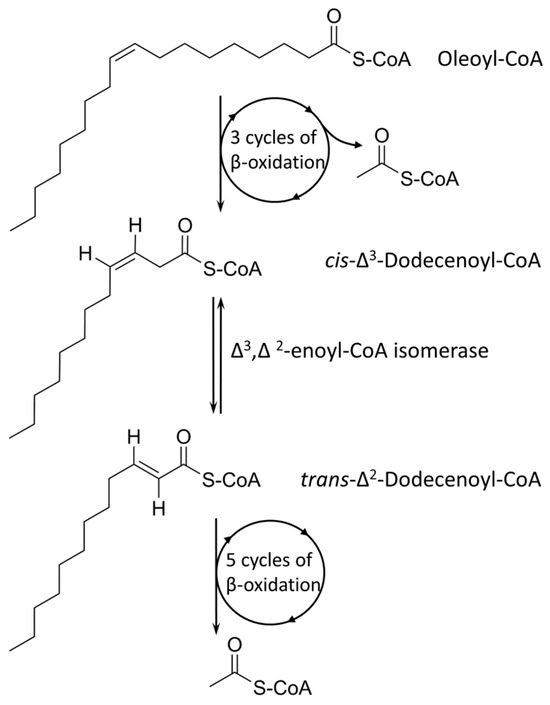{kind=link}
{kind=link}
{kind=link}
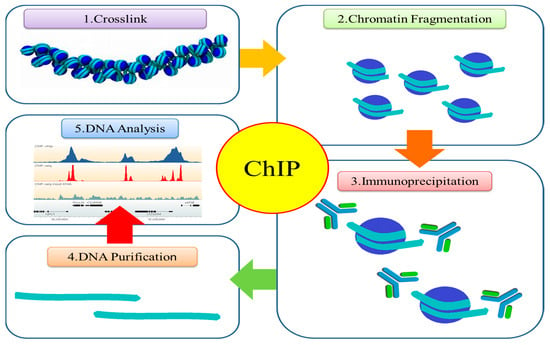{kind=link}
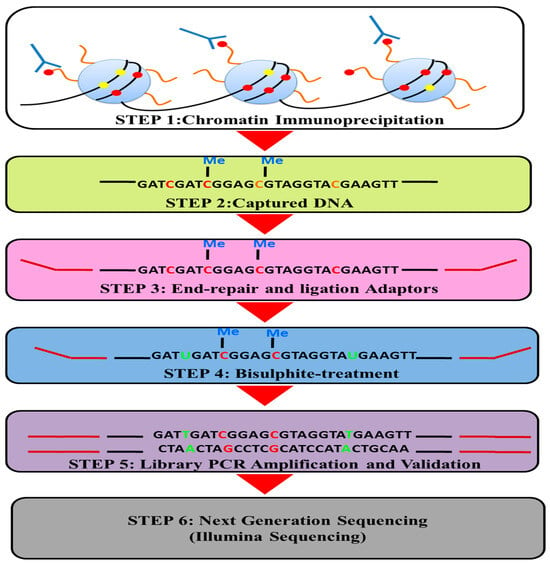{kind=link}
{kind=link}
{kind=link}
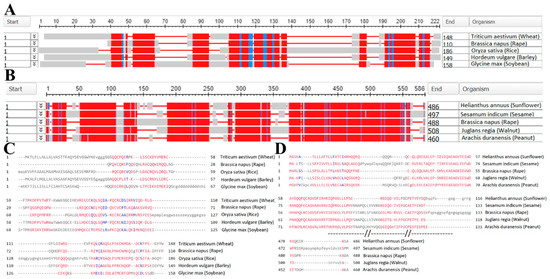{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}